
FDA recently issued updates to their Electronic Common Technical Document (eCTD) v4.0 Technical Conformance Guide; FDA eCTD v4.0 Module 1 Implementation Package. The package was issued without comment, and includes two components:
The Conformance Guide revision log states:
Revisions based on comments received during a public comment period.
Changes include removal of the following: 1) Two-way communications and associated data elements; 2) Regulatory Review Time; 3) Document media type, and 4) Category Event.
Although the document has been revised “based on comments”, there appears to have been no summary of comments or response to comments published in the Federal Register. As we shall see, the changes go well beyond what was mentioned in the log.
The most significant change was the removal of two-way communications with the FDA. The previous version of this document (V1.0, February 2020) had described a two-way communication process using the FDA’s electronic submission gateway (ESG). In fact, this exchange mechanism had been described since 2007 or earlier, when eCTD 4.0 (then known as Regulated Product Submissions or RPS) was first envisioned. The earlier document included the following process diagram.
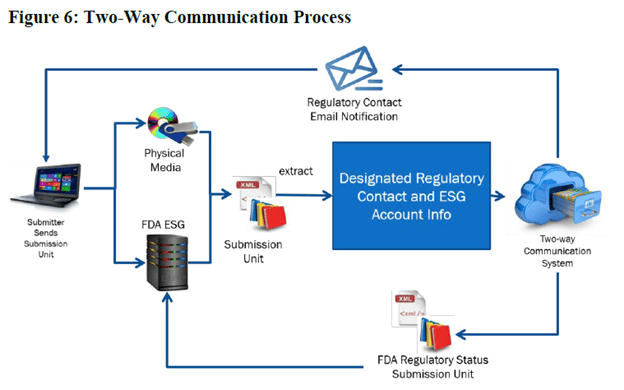
Since no rationale has been provided, we don’t yet know why this capability is being removed, but it does remove what has always been positioned as a major benefit of eCTD V4.0.
A new area that was added is Section 2.1.2 Patient Experience Data:
“If submitting patient experience data as part of an application for marketing approval, the following table should be populated and included in the Reviewer’s Guide (section 1.2). Patient experience data (e.g., clinical outcome assessments) collected as part of a clinical trial should be submitted as part of the relevant clinical trial data. Other patient experience data that is separate from clinical trials should be submitted to section 5.3.5.4.”
See the document for the specific table.
A number of other changes were made, most of which were not called out in the summary of revisions:
- Section 1.5.6 Use of Media Type was removed
- Section 1.6.1 Presubmissions: Advice on numbering was added. “Note: If a presubmission is transferred to eCTD v4.0, the next sequence number will be the next available sequence number as a whole number. For example, if transitioning after sequence number “0003”, the sequence following the transition mapping message will be sequence number “4”.”
- 1.6.3 Grouped Submissions: The previous discussion of file locations was removed: “The new files will physically reside in the primary application folder. When modifying files submitted in a grouped submission, it is possible that the referenced files are not located in the same application folder. The document identifier will be referenced if the file is not contained within the submitted content.”
- 1.6.3 Grouped Submissions: The following advice was added: “Consider standardizing keywords across applications so that you can form any grouped submission in the future. Keywords should be managed across application and all applications that belong to a group need to be transitioned prior to submitting a Grouped Submission in eCTD v4.0.”1.7.3
- 1.7.3 Contacts: the ESG Account Contact was removed.
- 1.7.5.1 Study Data Media Types was removed: “In eCTD v4.0, additional metadata is expected for study data files (e.g., CDISC SDTM, ADAM, SEND) by using media types specified in the FDA Module 1 eCTD v4.0 Implementation Package. FDA uses the media type to identify datasets for additional processing. If you do not include the media type, it may cause a delay in processing the submission and possible rejection of the sequence.”
- 1.8 Transition Mapping Message: clarified that “once an eCTD v4.0 submission is successfully received by the FDA, another transition mapping message cannot be submitted.”
- 2.1.6 Labeling (Section 1.14): under Labeling Samples, several paragraphs were removed: “Labeling documents may include both processing and support files. You should provide the relevant media type in the exchange message for each of the structured product labeling files. For the processing files (e.g., spl.xml), the code spl_proc should be applied to the document’s media type. For supporting files (e.g., image files), the code spl_supp should be applied to the document’s media type.
The media type in the message will be used to flag the content for further processing or specialized display.” - 2.1.8 Risk Evaluation and Mitigation Strategy (Section 1.16). This section was expanded to represent additional Module 1 headings that have been added for REMS, and to remove the requirement for a media type.
- 2.3.1 Lot Distribution Data: removed requirement for media type
- 5. Appendix A: Rules for Submission Tracking Information: no longer includes regulatory review times.
FDA Module 1 Electronic Common Technical Document (eCTD) v4.0 Implementation Guide v1.3 has also been published. This is a somewhat more technical document and notes the following changes:
Revised based on public comments from the “Electronic Common Technical Document v4.0 Technical Conformance Guide; Food and Drug Administration Electronic Common Technical Document v4.0 Module 1 Implementation Package; Request for Comments” Federal Register Notice. Changes include removal of the following: 1) Two-way communications and associated data elements; 2) Regulatory Review Time; 3) Applicant DUNS Number; 4) Document media type; and 5) Category Event. In addition, changes have been made to the folder structure and additional instructions for the placement of files for grouped submissions.
From the changes announced, one can question the overall direction and strategy FDA is taking towards the eventual genesis of eCTD and if there has been a re-evaluation of RPS in its current eCTD 4.0 guise. Rather than the more eye-catching eCTD expansion and development, the guidance seems to suggest a more practical, considered approach.
For some additional commentary on these changes, see my industry colleague Joel Finkle’s LinkedIn Post.