
1. Introduction: Why IDMP and xEVMPD Matter More Than Ever
If you work in regulatory affairs or operations in life sciences, you’ve likely encountered the terms IDMP and xEVMPD more times than you can count. And for good reason.

As global regulators demand more structured, standardized data to improve oversight, transparency, and safety, pharmaceutical companies are under increasing pressure to produce and manage a greater volume of product information. Health Authorities are also increasingly co-operating with one another, aiming to improve patient access and safety. That’s where IDMP and xEVMPD come in.
At their core, both frameworks are about making medicinal product data easier to submit, validate, share, and trust – across companies, borders, and regulatory bodies. xEVMPD serves as a central pharmacovigilance database for the European Union and EEA, ensuring that pharmacovigilance efforts in the EU could be coordinated by the EMA. IDMP goes further: its aim is to harmonize the exchange of medicinal product information worldwide. The European Medicines Agency (EMA) has led the way in implementing IDMP, but other health authorities will follow, expanding its reach globally.
For regulatory teams, this shift represents both a compliance challenge and a strategic opportunity. Those who can centralize and control their data effectively won’t just meet EMA requirements – they’ll be better positioned for long-term digital transformation and global IDMP roll-out.
In this article, we’ll walk you through everything you need to know – from the definitions of IDMP and xEVMPD, to the differences between them, EMA expectations, implementation challenges, and software solutions that can make your life a whole lot easier.
2. What is IDMP?
IDMP, or Identification of Medicinal Products, is a set of five ISO standards developed to improve the harmonization and exchange of medicinal product information worldwide. These standards ensure that medicinal products are described and identified using a consistent structure, regardless of who’s submitting the data or where.

The five ISO IDMP standards are:
• ISO 11615 – Medicinal product information
• ISO 11616 – Pharmaceutical product information
• ISO 11238 – Substances
• ISO 11239 – Information on pharmaceutical dose forms, units of presentation, routes of administration and packaging
• ISO 11240 – Units of measurement
Combined, these standards define how to describe a product’s:
• Ingredient substances and formulation
• Administration to a patient (route of administration, strength, and dosage form)
• Manufacturing and marketing authorization
• Packaging
• Regulatory and commercial status
• Clinical particulars
Why was IDMP created?
Regulators worldwide currently use disparate systems to track and review products. This makes it difficult to coordinate safety efforts across regions, detect global safety signals, or validate data across systems. IDMP enables a single source of truth for medicinal product data, making pharmacovigilance, regulatory submissions, and global collaboration far more efficient and accurate. While each individual regulator will implement IDMP slightly differently, IDMP will act as a ‘Rosetta stone’ for medicinal product information, allowing organizations to translate product information into the various global formats.
Where is IDMP required?
Currently, EMA is the first regulatory agency actively implementing IDMP in a phased approach through its SPOR data services. However, other agencies including FDA, Health Canada, and PMDA (Japan) are also working towards adopting IDMP in the future, signaling its move toward global standardization.
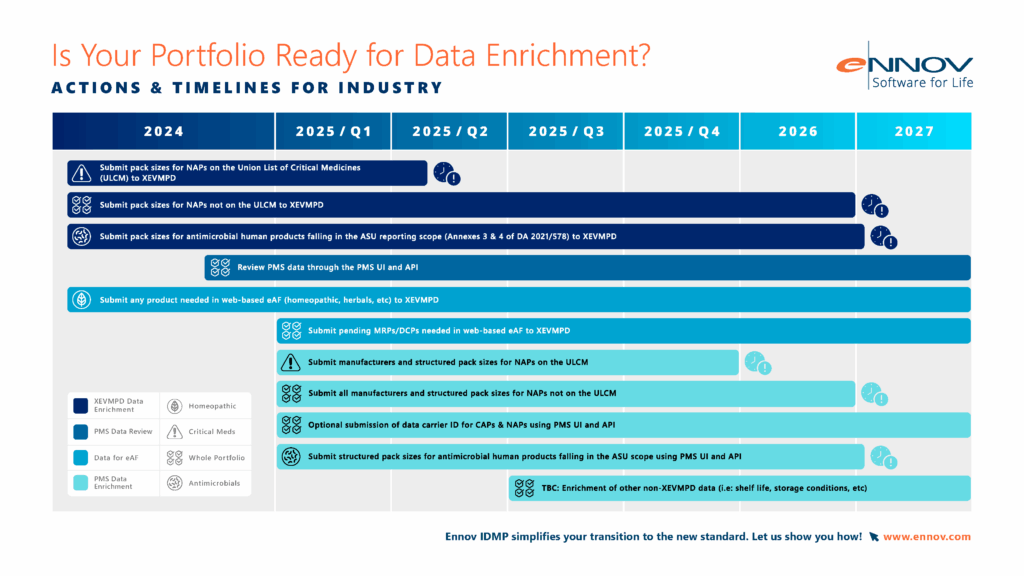
3. What is xEVMPD?
xEVMPD stands for Extended EudraVigilance Medicinal Product Dictionary, and it’s a European data submission standard that predates IDMP. First made mandatory in 2012, it now represents a first step toward structured product data, designed to support pharmacovigilance and post-market surveillance in the EU.
xEVMPD requires marketing authorization holders (MAHs) to submit product information to the EudraVigilance database. These submissions include data on ingredients, strengths, dosages, route of administration, and regulatory status – albeit in a less complex and structured format than IDMP.
Why xEVMPD was introduced
The EMA introduced xEVMPD in order to fulfill Directive 2010/84/EU, which expanded pharmacovigilance responsibilities and aimed to improve the early detection of adverse events. By collecting structured data through the EVWEB portal, regulators gained faster access to medicinal product information across all EU countries.
What are the limitations of xEVMPD?
While xEVMPD has been foundational for EMA’s pharmacovigilance infrastructure, it has several drawbacks:
- Limited data granularity compared to IDMP
- Inconsistent terminology due to lack of controlled vocabularies
- Manual entry errors through the EVWEB interface
- Limited scalability for large-scale, multi-country operations
XEVMPD remains a key part of the European pharmacovigilance network, and makes the case for a robust, Europe-wide data sharing framework – which brings us to IDMP.
4. IDMP vs xEVMPD: Key Differences and Evolution
If you’re already handling xEVMPD submissions, you might wonder why health authorities think IDMP is necessary at all. The short answer: IDMP is more structured, more powerful, and future-ready.
While both frameworks serve a similar purpose – improving the quality and accessibility of medicinal product data – they differ significantly in scope, structure, and ambition.
Comparison: IDMP vs xEVMPD
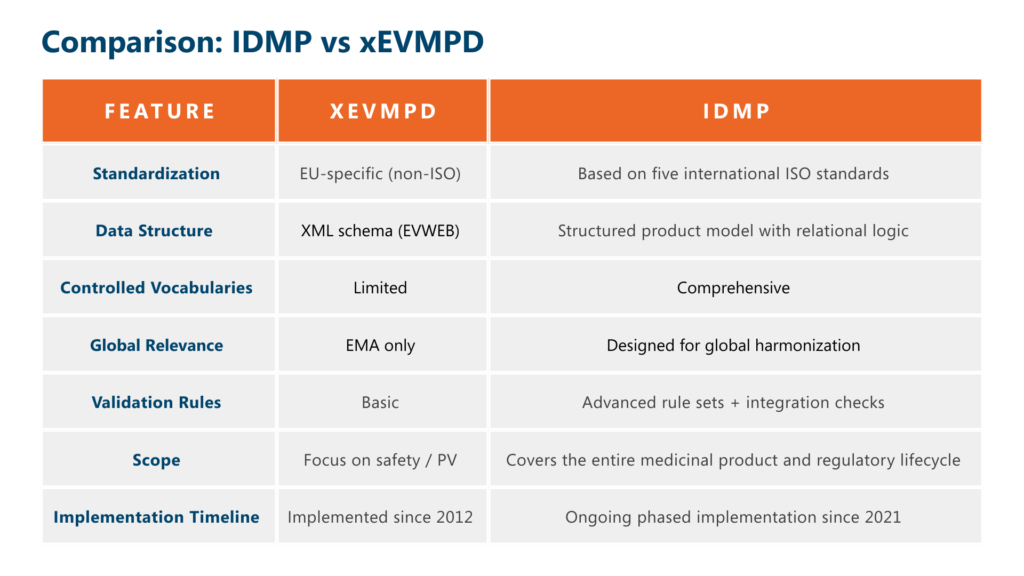
So what does this mean for you?
If you’re already compliant with xEVMPD, you’re on the right track – but it’s not enough. IDMP builds on xEVMPD, adds complexity and depth, and introduces far more rigorous requirements for data quality, traceability, and integration.
EMA has made it clear that IDMP is the future. While both frameworks are in play today, IDMP will eventually replace xEVMPD for most submission types.
5. Understanding EMA’s Requirements for IDMP & xEVMPD
The EMA is rolling out IDMP through a phased implementation plan centered around its SPOR model – Substance, Product, Organisation, and Referential data.
If you’re submitting to EMA (or plan to), understanding the expectations and timelines is key to avoiding delays or non-compliance.
Key EMA resources
1. SPOR Services
SPOR went live in 2017, and is accessed through the SPOR Portal. You’ll need to ensure your internal data aligns with these domains.
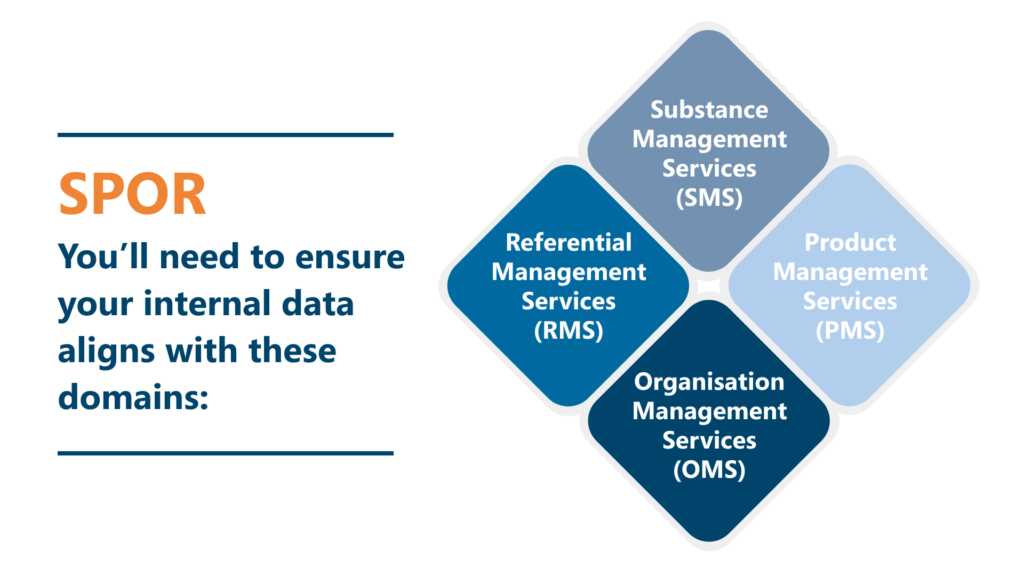
• Substance Management Services (SMS)
• Product Management Services (PMS)
• Organisation Management Services (OMS)
• Referential Management Services (RMS)
2. EU Implementation Guides
This document outlines the specific rules for data format, validation, and submission under IDMP. It’s mandatory reading for regulatory and IT teams involved in implementation.
The guide is broken down into chapters covering:
- Chapter 1: Registration requirements – guidance on getting to SPOR
- Chapter 2: Data elements for the electronic submission of information on medicinal products for human use – guidance on which medicinal product information will need to be submitted under the EMA’s IDMP guidelines
- Chapter 3: Process for the electronic submission of medicinal product information – guidance on the process for submission
- Chapter 4: Data quality assurance
- Chapter 5: Data access/export – guidance on access and privileges established for PMS users
- Chapter 6: Technical specifications on structure and format – technical specifications for the SPOR APIs, including implementation guides
- Chapter 7: Migration guide – guidance on how data is migrated from XEVMPD and the EMA’s internal product database, SIAMED, including backwards compatibility rules
- Chapter 8: Practical examples – examples of how to represent medicinal product information in the EMA IDMP format and in PMS
- Chapter 9: Process for submitting existing data on medicinal products authorised for human use – SIAMED and XEVMPD deltas
6. Data Requirements: What Structured Data Do You Need to Manage?
Let’s get practical. IDMP and xEVMPD both require companies to collect, organize, and submit structured data about their medicinal products – but IDMP raises the bar significantly.
What kind of data does EMA expect under IDMP?
The IDMP standards define hundreds of specific data fields, many of which require linking across domains (e.g., product + substance + manufacturer). All of this needs to be machine-readable, validated against controlled vocabularies, and easily traceable back to source systems.
EMA is implementing a phased approach to IDMP: some types of information are already managed in an IDMP-compliant framework, such as organisation information through OMS. Other data will need to be enriched iteratively in PMS, in alignment with SMS, OMS and RMS.
How does this differ from xEVMPD?
• xEVMPD required far less granular data – e.g., a product’s substance name but not its full substance composition or source.
- xEVMPD doesn’t require as much information in structured formats – e.g pack sizes are submitted as free text, rather than structured data fields
• IDMP forces companies to think about data interoperability, traceability, and long-term master data quality.
Where do most companies struggle?
• Finding the data: It’s often scattered across Regulatory, Quality, and Supply Chain systems.
• Structuring it correctly: Most legacy systems aren’t built for IDMP’s level of detail.
• Maintaining updates: IDMP compliance isn’t a one-time task – it requires continuous data stewardship.
7. Common Challenges with IDMP/xEVMPD Compliance
No regulatory professional needs to be reminded that compliance is rarely straightforward – and when it comes to IDMP and xEVMPD, the complexity is real.
Despite the long runway provided by EMA’s phased implementation, many life sciences companies still struggle with the transition. That’s because these standards don’t just require a new way of submitting data – they require a whole new way of thinking about data.
Here are some of the most common challenges organizations report.
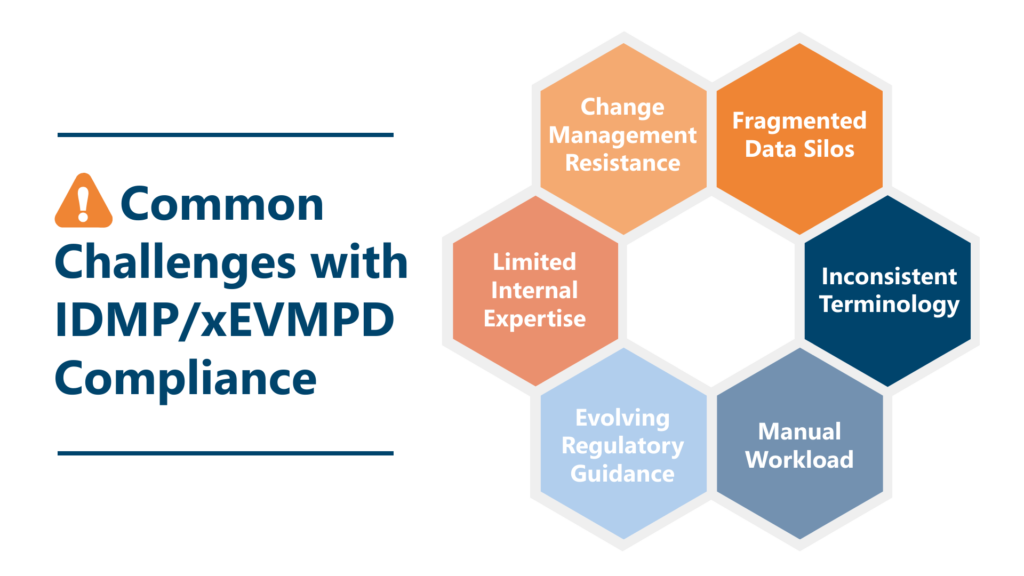
1. Fragmented Data Silos
Product data is often scattered across multiple departments – regulatory, manufacturing, supply chain, quality – and stored in different formats, systems, and spreadsheets. Without centralized access, gathering the required data becomes a scavenger hunt.
2. Inconsistent Terminology
IDMP mandates the use of controlled vocabularies from EMA’s SPOR Referential, which means internal naming conventions must be mapped (and often reworked) to align with standard terms.
3. Manual Workload
Legacy processes typically involve manual data entry into EVWEB (for xEVMPD) or spreadsheets for IDMP prep – both of which are error-prone, resource-intensive, and unsustainable as submission volumes grow.
4. Evolving Regulatory Guidance
EMA has refined the Implementation Guides over time, and companies that started early regularly find they need to revisit previous work to stay compliant.
5. Limited Internal Expertise
IDMP is deeply technical. Many regulatory teams lack in-house experts who understand both the ISO standards and the underlying data architecture required for compliance.
6. Change Management Resistance
Operational change – especially in global organizations – can be slow. Getting buy-in from cross-functional stakeholders is a challenge, especially when the immediate ROI isn’t obvious.
8. How to Prepare: Building a Strong Data Governance Foundation
If there’s one theme that comes up in every successful IDMP implementation story, it’s data governance.
Meeting EMA requirements isn’t just about ticking boxes. It’s about setting up systems, roles, and rules to manage medicinal product data in a consistent, high-quality, and traceable way – now and in the future.
Here’s how to lay the foundation:
1. Establish Ownership
Create a cross-functional IDMP task force. This should include Regulatory Affairs, IT, Quality, Supply Chain, and Master Data Management teams. Assign clear roles for who owns what data and clear processes for managing the lifecycle of each data point.
2. Perform a Data Gap Analysis
Audit your existing data against EMA’s IDMP requirements, focusing on the early phases of the EMA’s iterative data requirements. Where are the gaps? Is data missing, outdated, or stored in non-structured formats?
3. Harmonize Internal Terminology
Start aligning product attributes with EMA’s controlled vocabularies – especially for dose forms, routes of administration, units of presentation, and packaging types.
4. Centralize Your Data
Adopt a single source of truth approach. Whether it’s a Regulatory Information Management (RIM) system or a purpose-built IDMP tool, having centralized, version-controlled data is critical.
5. Automate Where Possible
Look for software that can validate your data against SPOR vocabularies, generate IDMP-compatible output formats (e.g., HL7 FHIR), and support submission directly to EMA systems.
6. Train Your Teams
Provide targeted training for each group involved in the IDMP process. Regulatory should understand SPOR and submission logic; IT should know integration protocols; and data stewards must learn validation workflows.
9. The Role of SPOR in IDMP and xEVMPD

You can’t talk about IDMP without talking about SPOR. This is EMA’s master data framework – and it’s the engine powering structured data submissions under both IDMP and xEVMPD.
What is SPOR?
SPOR stands for:
• Substance Management Services (SMS)
• Product Management Services (PMS)
• Organisation Management Services (OMS)
• Referential Management Services (RMS)
These services manage all reference and master data that companies need to submit valid structured product information.
Why is SPOR critical?
Because SPOR is the gatekeeper. Data such as substances, organisations and referentials can only be selected from the SPOR services: it’s simply not possible to submit IDMP data to EMA without using SPOR.
For example:
• Organisation names and locations must be pre-registered in OMS.
• Product submissions must use terms from RMS (e.g., dose form presentations such as “film-coated tablet” must match EMA’s official entry).
• Substance data must be aligned with SMS entries for active ingredients and excipients.
How to Work with SPOR
• Use EMA’s SPOR portals to search for and download vocabularies.
- Map your data to SPOR as appropriate.
• Set up API integrations to keep your RIM or IDMP system in sync with SPOR updates.
• Validate your entries before submission to minimize rejection risk.
Where do companies get tripped up?
• Not mapping each organisation to an OMS organisation and location.
• Using legacy internal terms that don’t match SPOR, leaving them unable to submit this data.
• Failing to monitor SPOR updates, which can affect validation rules.
Being SPOR-ready is one of the most practical ways to de-risk your IDMP and xEVMPD compliance efforts.
10. How IDMP Compliance Software Helps
Trying to manage IDMP and xEVMPD requirements manually is like trying to file your taxes with a pencil and a Rolodex. Technically possible, sure. But slow, risky, and increasingly unsustainable.
That’s where IDMP compliance software steps in.
What does IDMP software do, exactly?
At a high level, a modern IDMP compliance tool helps you:
• Centralize product data from multiple departments and sources
• Validate your data against EMA’s controlled vocabularies and SPOR
• Generate submission-ready outputs in the required formats (e.g., IDMP HL7 FHIR or xEVMPD XML)
• Track data lineage for audit purposes and change management
• Automate updates to reflect regulatory changes or product variations
Key Features to Look For in IDMP software
Not all software is created equal. When evaluating IDMP solutions, look for tools that offer:
• Native SPOR integration (OMS, RMS, PMS APIs)
The ability to compare your data to EMA’s PMS database
• Multi-region and multi-language support
• Version control for every product data element
• Analytics and reporting dashboards
Software solutions like Ennov’s IDMP EASI are purpose-built for regulatory professionals, allowing you to manage xEVMPD and IDMP submissions in a unified environment — not in separate silos.
Why it matters
Compliance software isn’t just a “nice to have” — it’s what enables speed, accuracy, and scalability.
With regulatory authorities moving toward automated validation and structured data exchange, manual workflows simply won’t cut it.
11. Submission Workflows: How xEVMPD and IDMP Submissions Work
Let’s get into the mechanics of it. While both xEVMPD and IDMP require structured submissions, they differ significantly in terms of technology, process, and format.
xEVMPD Submissions
• Platform: Submissions are made via EVWEB, the EMA’s web-based portal for EudraVigilance data, or via a gateway within your RIM tool.
• Format: Data is submitted in XML format, structured according to the xEVPRM schema.
• Validation: EVWEB validates data in real-time and may return errors requiring correction before submission is accepted.
• Use cases: Required for all MAs in the EU and EEA.
IDMP Submissions (Current and Future State)
• Platform: EMA will transition to structured data exchange via the Product Management Service (PMS).
• Format: IDMP data is expected to be submitted using the HL7 FHIR standard, a modern, flexible health data model.
• Integration: Submissions will be made through API-based exchange or via the PMS user interface.
• Validation: Automated validation using SPOR and rule sets from the EU Implementation Guide.
• Scope: Initially applied to medicines on the Union List of Critical Medicines, but expanding in scope to cover all products authorised in the EU and EEA.
Key Transition Considerations
• xEVMPD is still required in the near term, even as IDMP is being implemented.
• EMA will gradually shift to IDMP-only structured data for all regulated products.
• Companies must prepare now to move from portal-based to system-to-system submission models.
12. Best Practices for Successful IDMP/xEVMPD Projects
Whether you’re starting from scratch or have already been working on your IDMP implementation, the same core principles apply. The most successful companies follow a structured, phased approach that combines technology, data quality, and cross-functional ownership. Here are proven best practices:
1. Start Early and Small
Now that there are EMA deadlines, it’s time to start moving. Begin with a pilot: pick a few products and a prioritized scope, and run a full data extraction, validation, and submission simulation.
2. Treat It as a Business Transformation
IDMP isn’t just a regulatory checkbox — it’s a data governance initiative. Treat it like an enterprise program, not a side project.
3. Secure Executive Sponsorship
You’ll need support across departments, which often means you’ll need buy-in from leadership. Make the case for how better data supports broader goals: speed to market, reduced compliance risk, and digital maturity.
4. Create Reusable Data Models
Avoid “one-off” submissions. Build repeatable processes and templates that can scale across product families and markets.
5. Integrate with Existing Systems
Link your IDMP solution with existing RIM, ERP, and document management platforms to avoid duplication and reduce manual errors. Avoid manual processes (for example, using Excel to gather data).
6. Monitor Regulatory Updates
EMA frequently updates guidance documents. Subscribe to updates, attend webinars, and stay close to industry working groups.
7. Focus on Data Quality from Day One
Bad data = failed submissions, data rework and lost time. Invest early in validation, cleanup, and process controls that ensure data stays compliant over time.
13. The Future of IDMP: Global Alignment and Automation

The Identification of Medicinal Products (IDMP) standards are not just a European initiative; they represent a global movement towards harmonized medicinal product data. As regulatory agencies worldwide recognize the benefits of standardized data for patient safety and regulatory efficiency, the future of IDMP is poised for broader adoption and increased automation.
Global Adoption Trends
While the European Medicines Agency (EMA) has been at the forefront of IDMP implementation, other regulatory bodies are following suit:
• United States: The Food and Drug Administration (FDA) has initiated efforts to align with IDMP standards, particularly in areas like substance identification and pharmacovigilance reporting.
• Canada: Health Canada is exploring the adoption of IDMP to enhance regulatory submissions and product tracking.
• Japan: The Pharmaceuticals and Medical Devices Agency (PMDA) is participating in global discussions to integrate IDMP standards into its regulatory framework.
This global momentum underscores the importance for pharmaceutical companies to adopt IDMP-compliant systems that can cater to multiple regulatory jurisdictions.
Advancements in Automation
The transition to IDMP is closely linked with the push towards automation in regulatory processes:
• Data Exchange: The adoption of HL7 FHIR (Fast Healthcare Interoperability Resources) facilitates seamless data exchange between stakeholders, reducing manual interventions and errors.
• Artificial Intelligence (AI): AI-powered tools are being developed to assist in data mapping, validation, and submission processes, enhancing efficiency and accuracy.
• Integration with Product Lifecycle Management (PLM): Aligning IDMP data with PLM systems ensures that product information remains consistent and up-to-date throughout its lifecycle.
Embracing these technological advancements will not only aid in compliance but also position companies for operational excellence in the digital age.
14. FAQs on IDMP & xEVMPD
Navigating the complexities of IDMP and xEVMPD can raise numerous questions. Here are some frequently asked questions to provide clarity:
1. What is the primary difference between IDMP and xEVMPD?
While both aim to standardize medicinal product data, xEVMPD is an EU-specific system primarily focused on pharmacovigilance, whereas IDMP is a set of global ISO standards designed for comprehensive product identification and data harmonization.
2. Is xEVMPD still relevant with the advent of IDMP?
Yes, xEVMPD remains in use. However, the EMA is transitioning towards IDMP standards, and it’s anticipated that IDMP will eventually supersede xEVMPD.
3. How does SPOR relate to IDMP?
SPOR (Substance, Product, Organisation, and Referential) is the EMA’s master data initiative that underpins IDMP implementation by providing standardized data services essential for compliance.
4. How often does EMA update IDMP guidelines?
The EMA periodically updates its guidelines to reflect evolving standards and stakeholder feedback. It’s crucial to monitor the EMA’s official communications for the latest information.
5. Can small and medium-sized enterprises (SMEs) manage IDMP compliance effectively?
Yes, SMEs can achieve compliance by leveraging scalable IDMP solutions tailored to their needs and by seeking guidance from regulatory consultants specializing in IDMP.
6. Are there industry collaborations to facilitate IDMP implementation?
Various industry consortia and working groups collaborate to share best practices, develop tools, and provide support for IDMP implementation across the pharmaceutical sector.
15. Conclusion: Turning Compliance into Competitive Advantage
Achieving compliance with IDMP and xEVMPD is more than a regulatory obligation; it’s an opportunity to enhance data integrity, streamline operations, and improve patient safety. By proactively adopting these standards, pharmaceutical companies can:
• Enhance Regulatory Efficiency: Standardized data facilitates smoother interactions with regulatory agencies, expediting approvals and reducing queries.
• Improve Data Quality: A structured approach to data management minimizes errors and inconsistencies, leading to better decision-making.
• Foster Innovation: Reliable data serves as a foundation for advanced analytics, AI applications, and personalized medicine initiatives.
In essence, embracing IDMP and xEVMPD compliance transforms a regulatory challenge into a strategic asset, positioning companies for success in an increasingly data-driven healthcare landscape.